New approach to ab initio modelling of electron-phonon interactions in correlated materials
by Carey Sargent, EPFL, NCCR MARVEL
While first-principles approaches allow for the accurate calculation of electron-phonon (e-ph) interactions in a wide range of solids, density-functional theory often fails to describe the ground state in correlated electron systems (CES). This has ruled out reliable e-ph calculations for materials including transition metal oxides, high-temperature superconductors and multiferroics, hindering theoretical study.
Now, in the Letter “Ab initio electron-phonon interactions in correlated electron systems,” just published in Physical Review Letters, researchers have presented an accurate approach to studying these very interactions. The novel method captures the interplay of entangled charge, spin and phonon degrees of freedom, and can describe subtle aspects such as how the localized electrons and their spin interact and are affected by atomic vibrations.
The method, set to pave the way for more accurate studies of correlated materials such as high-temperature superconductors, planetary materials, multiferroics and complex oxides, is the fruit of synergies between Professor Nicola Marzari’s group at EPFL and that of Professor Marco Bernardi at Caltech. Their respective expertise in two cutting-edge computational tools — Hubbard-U corrected density-functional theory and electron-phonon interactions — was essential in developing the new method.
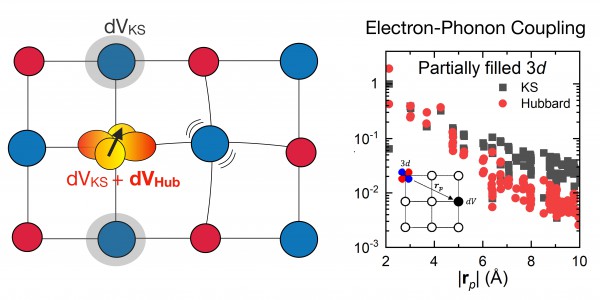
Left: In correlated materials, the localized electrons occupying d atomic orbitals (shown in yellow) couple with the atomic vibrations (phonons) in an unconventional way – via perturbation of the localized electronic repulsion, the so-called Hubbard term.
Right: In cobalt oxide, a correlated Mott insulator, this Hubbard-mediated electron-phonon coupling is as strong as the conventional coupling due to the mean-field Kohn-Sham (KS) potential.
Hubbard-U
While e-ph interactions in correlated materials can be obtained through a combination of density-functional theory (DFT) to compute the electronic structure and density-functional perturbation theory (DFPT) to characterize the lattice dynamics, quantitative study remains challenging. This is because standard DFT usually fails to correctly describe their ground state.
In DFT, electronic interaction energies are described as the sum of classical repulsion between electronic densities and a term meant to cover all correlations and spin interactions. It is difficult to model the dependence of this term, the xc functional, on electronic charge density and it is not suited to capturing many-body features of certain ground states. It does not, for instance, describe correlated systems, with physical properties controlled by many body electronic interactions, very well. The functional tends to introduce self-interaction errors in certain localized electrons and fails to correctly capture the strong e-ph coupling and polaron effects seen in correlated transition metal oxides (TMOs).
A solution has been proposed in the Hubbard correction, U, which treats the strong on-site interaction of localized electrons with an additional term, while the rest of the valence electrons are approached through normal DFT approximations. The resulting DFT+U model is particularly suited to counteracting the self-interaction error of DFT and it can also be computed ab initio—the framework is then free of empirical parameters. DFT + U’s linear response variant DFPT + U, has been used to study the electric structure and lattice dynamics of many TMOs.
For these reasons, the researchers chose a framework of DFT + U and DFPT + U for their first-principles calculations of e-ph interactions in CES. In the Letter, they applied their approach to cobalt oxide, a prototypical Mott insulator. Such materials are expected to conduct electricity according to conventional band theory but turn out to be insulators. The researchers showed that while DFT alone predicts it to be a dynamically unstable metal, their approach correctly predicts its insulating ground state.
The detailed investigations of the e-ph interactions and electron spectral functions in cobalt oxide allowed them to show how their approach is one that is broadly applicable and affordable for quantitative studies of e-ph interactions in CES and provides a novel theoretical tool for interpreting experiments in this broad class of materials.
Reference:
Jin-Jian Zhou, Jinsoo Park, Iurii Timrov, Andrea Floris, Matteo Cococcioni, Nicola Marzari, Marco Bernardi, Ab Initio electron-phonon interactions in correlated electron systems, Physical Review Letters (2021).
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter